
The Membrane Potential Paradox: How Bacteria Balance Permeability and Energy
Published:
The Membrane Potential Paradox: How Bacteria Balance Permeability and Energy
It took longer that I thought, but I’m thrilled to share that the core of this work—my main thesis—is now out in Nature Microbiology (link to full paper here). It’s surreal to move from debugging leaky microfluidics chips at 6 a.m. to seeing the story published. Now, let me tell you what it is about.
The leak in the wall
In the world of Gram-negative bacteria, the outer membrane serves as a protective wall, guarding the cell from external threats. Yet like all great walls, it has gates, these are protein channels called porins, that mediate the influx of essential nutrients for the cell and, incidentally, also antibiotics. Since the early studies of porins by Nikaido in the 1980s, these gates were largely considered to be wide open, allowing free passage for small molecules. This assumption, however, presented a fundamental paradox. Every biologist knows that bacteria rely on the proton motor force, that is, the accumulation of a proton gradient in the periplasmic space, as a source for ATP synthesis, motility, and transport. Thus, if porins as always open, then protons can simply leak out through these open gates? This question of energy conservation versus nutrient access led us to investigate what we call the membrane potential paradox.
Following the sugar
Our first step was to watch things flow. We used a fluorescently-tagged glucose molecule (2-NBDG), as a tracer to see how much was getting into the cells. As you’d expect, when we deleted the genes for the main porin proteins (ompF, ompC, ompG, nmpC, phoE), uptake dropped. No gates, no traffic. But then we got our first surprise. While screening a library of knockout mutants, we found one that had nothing to do with porins but still blocked uptake: a mutant for a potassium channel called Kch. [It was a strange and exciting clue.] This pushed us to think about ions. Maybe the porins weren’t just passive pores but were instead sensitive to the ionic environment. We tested changing the pH and potassium levels outside the bacteria, but nothing happened. However, we reasoned, maybe it’s the inside that matters. We used chemicals called ionophores such as CCCP, which makes the membrane leaky to protons, and valinomycin, which does the same for potassium to mess with the periplasmic ion concentrations directly. And it worked! We saw a huge change in the uptake of our fluorescent tracer. This was a big moment, suggesting that porins were indeed regulated by internal ions. But as a scientist, you’re trained to be skeptical of your own results. Ionophores are messy; they can have all sorts of side effects. We struggled with this idea. Using ionophores could have second-order effects. The solution, funnily enough, came from a completely different field: neuroscience. Neuroscientists care deeply about membrane voltage because neural is key for understanding neuron physiology. Thus, they developed this amazing toolbox of light-activated proteins that can be used to control the flow of ions across the membrane using light. We found one called ArchT, a proton pump originally discovered in algae, that you can switch on with a flash of green light. The thought was simple: what if we put this pump into our bacteria and used light to pump protons directly into the periplasm? We set up our bacteria in a microfluidic chip under the microscope. In the dark, the cells were happily swallowing up the fluorescent glucose. Then, we flipped the switch for the 561 nm laser. And just like that, the uptake stopped (Figure 1).
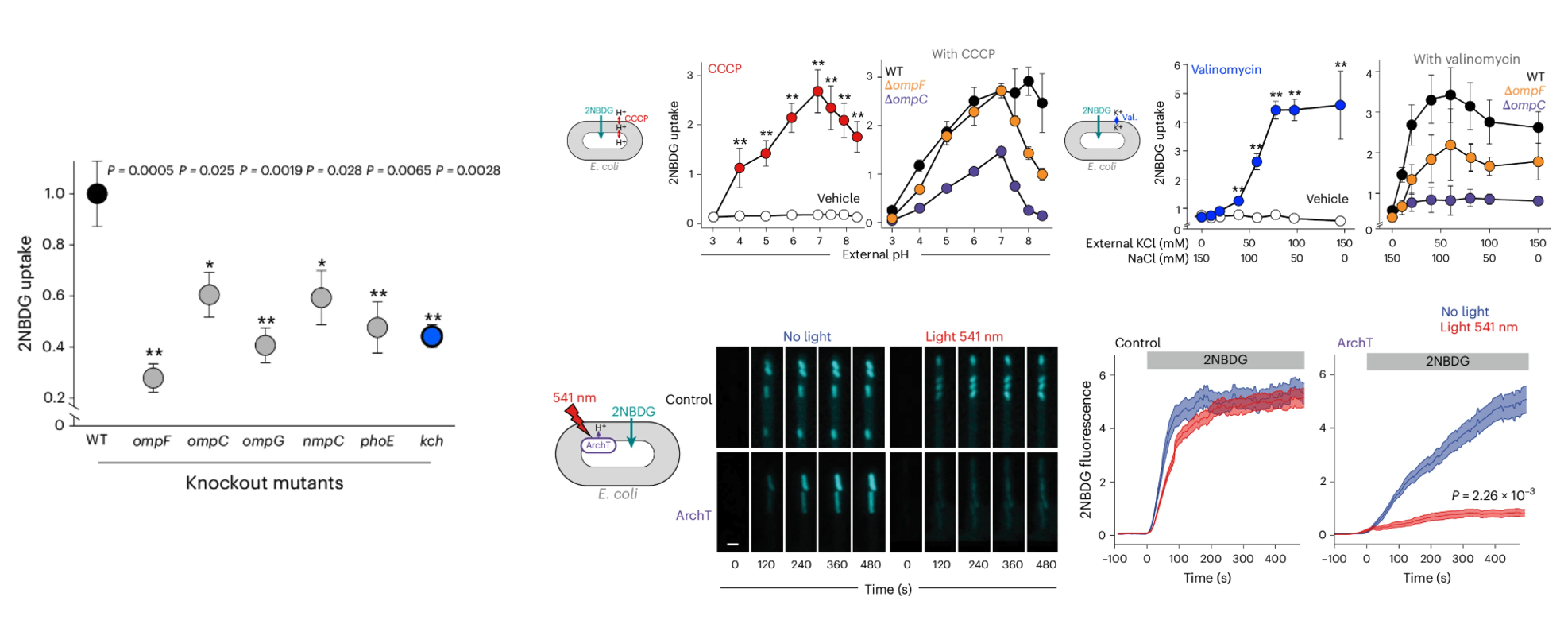
Figure 1. Internal H+ and K+ reshape porin permeability and light-driven ArchT pumping rapidly blocks 2NBDG uptake.
Where the pore flexes
Now we had proof, but we needed a mechanism. Previous literature had focused on the inside L3 loop of porins because it formed a constriction zone. But our molecular dynamics simulations, run by a collaborator, pointed elsewhere; they suggested that protonating charged residues on the periplasmic side for neutral residues (E23A, K27A, D28A, E64A, D69A, D156A, D162A, D289A, K329A) of the porin would cause it to constrict. This was a new idea. To test it, we spent months creating an OmpC mutant where we replaced all those charged residues with neutral alanines. The phenotype of this mutant was the perfect confirmation: it was leakier than wild-type and, crucially, had lost its ability to be regulated by pH (Figure 2).
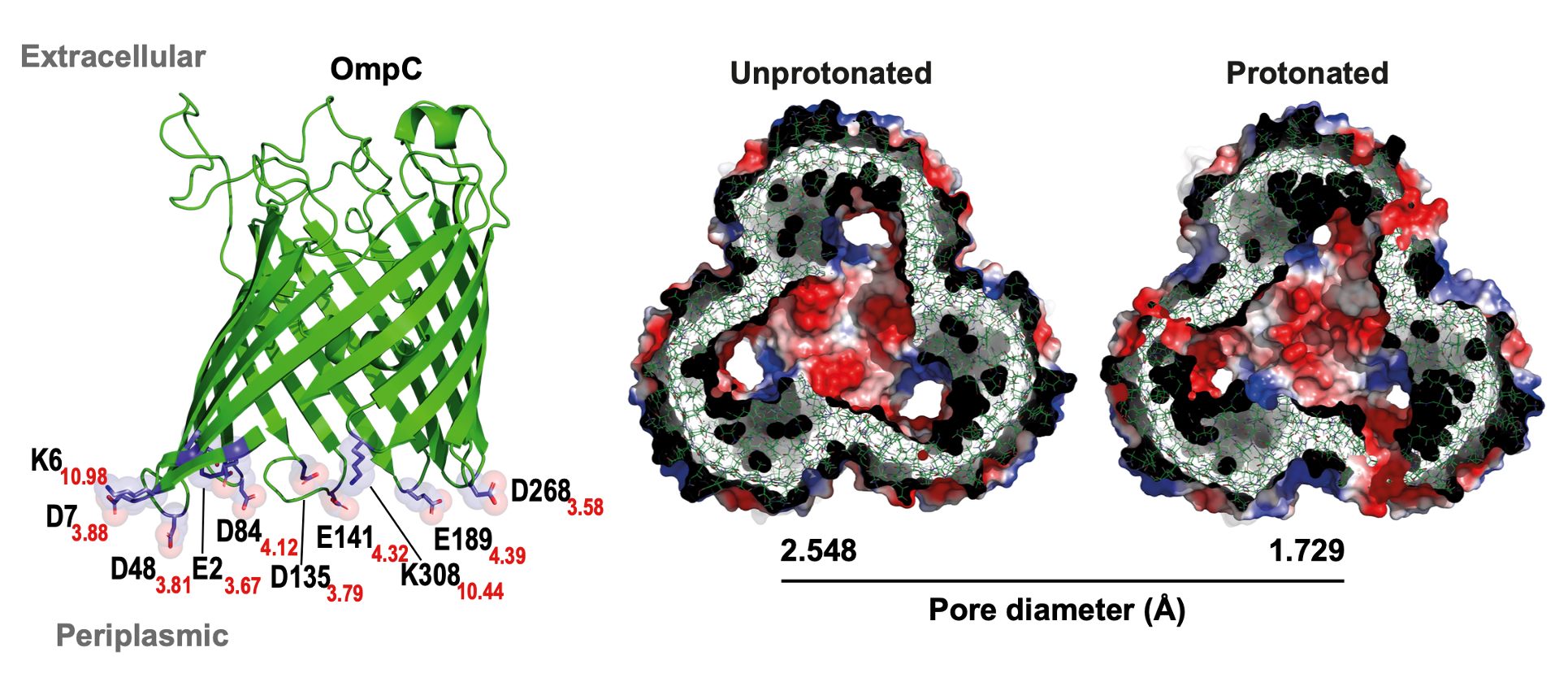
Figure 2. Periplasmic protonation of conserved charged residues narrows the OmpC pore, explaining the loss of regulation in charge-neutral mutants.
The metabolic engine
It was now logical to ask: Does bacterial physiology have a mechanism to carry out this kind of regulation spontaneously? To find out, we adapted a wide range of genetically encoded fluorescent sensors (pHluorin, pHuji, GcaMp, and Ginko1) to monitor ion concentrations. Our most significant innovation was adapting these sensors for the periplasmic space. We initially thought that exporting them with pelB tags was sufficient, but quickly discovered that proteins do not tolerate the highly oxidative environment of the periplasm. For pHluorin, we found the dynamic range was not wide enough, so we switched to pHuji, which covers lower pH values. For Ginko1, we had multiple failed attempts because it relied on a GFP backbone anchored with a potassium-binding region. The issue with GFP is that it has disulfide bonds that do not form properly in the periplasmic space. Thus, we had to change the GFP for sfYFP while maintaining the potassium-binding region. With these tools, we plugged the cells into our microfluidic devices and began monitoring ion dynamics. We observed spontaneous spikes in membrane potential—bacterial “action potentials,” if you wish—that were mirrored by changes in periplasmic H+ and K+. Crucially, these action potentials were absent in the Kch knockout cells. The most exciting part came when we simultaneously imaged the membrane voltage alongside 2NBDG uptake. This revealed that moments of membrane depolarization correlated perfectly with bursts of 2NBDG uptake (Figure 3). Our mental model to explain this dynamic resembled a 4-piston engine cycle. The cycle begins in the “power stroke” or hyperpolarized state, with the periplasmic space full of protons, representing stored potential energy. These protons are then rapidly consumed to produce ATP or power other biological processes (Exhaust). As the proton gradient is consumed, the membrane begins to depolarize. This drop in potential acts as the “intake valve opening” (fuel intake), allowing nutrients (or 2-NBDG) to rush in through the now-open porin gates. Finally, the bacteria immediately triggers another metabolic cycle (Compression) to pump protons back into the periplasm, re-hyperpolarizing the membrane and closing the porin gates, ready to fire the next “power stroke.” Returning to our original claim that bacteria use the PMF as a source of energy, we tested how different carbon sources would impact these bacterial “action potentials.” We provided the bacteria with sources that would lead to different levels of membrane potential. Interestingly, we observed a higher frequency of membrane voltage oscillations when E. coli was supplied with glucose compared with other carbon sources such as fumarate or lipids. More importantly, the observed synchrony, depolarization correlating with uptake and hyperpolarization with low uptake, was consistent. This was a fascinating discovery suggesting that bacteria could be using these membrane voltage oscillations to dynamically regulate the uptake of small molecules (Figure 4).
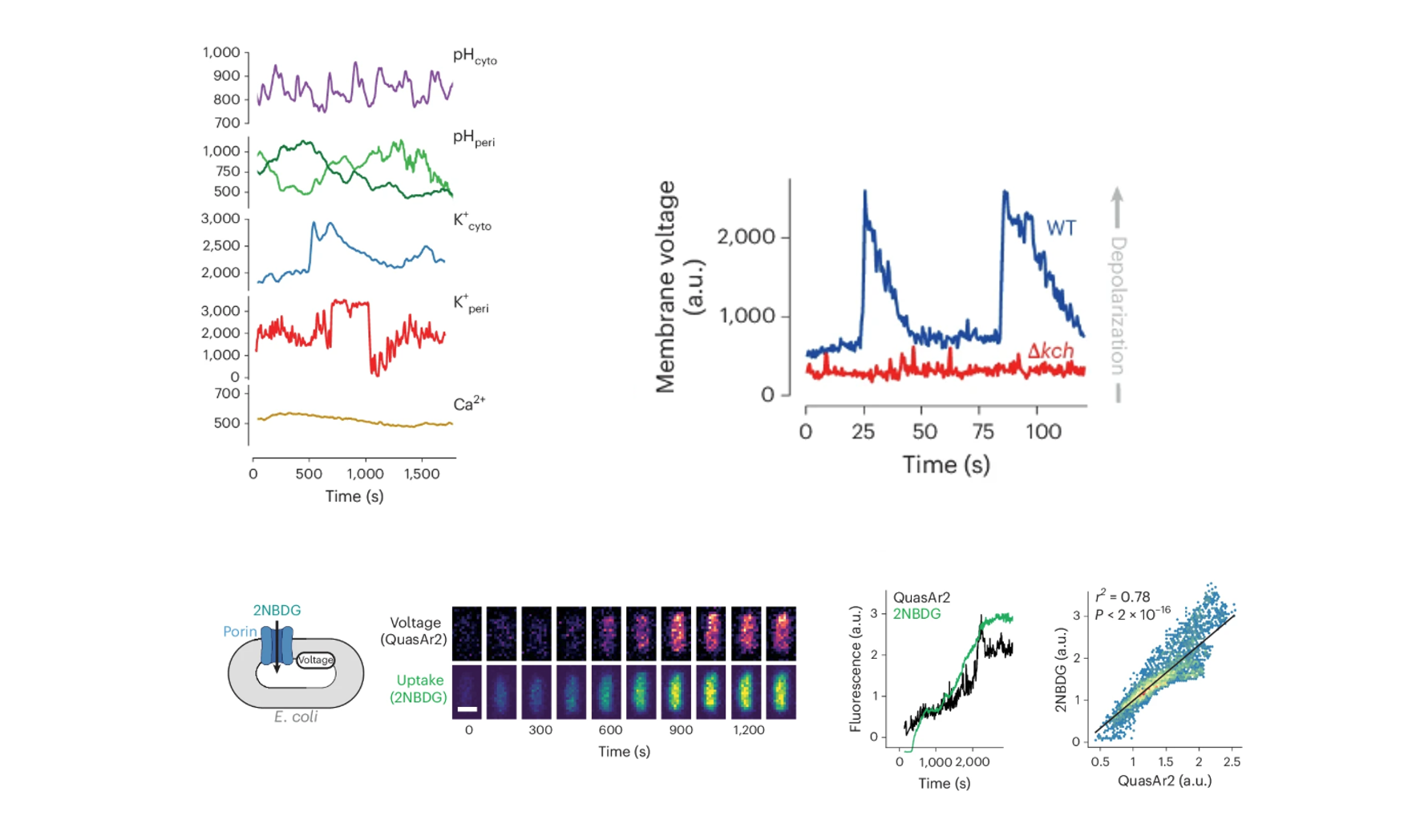
Figure 3. Ion-sensor imaging links periplasmic H+/K+ fluctuations with membrane depolarization and bursts of 2NBDG uptake.
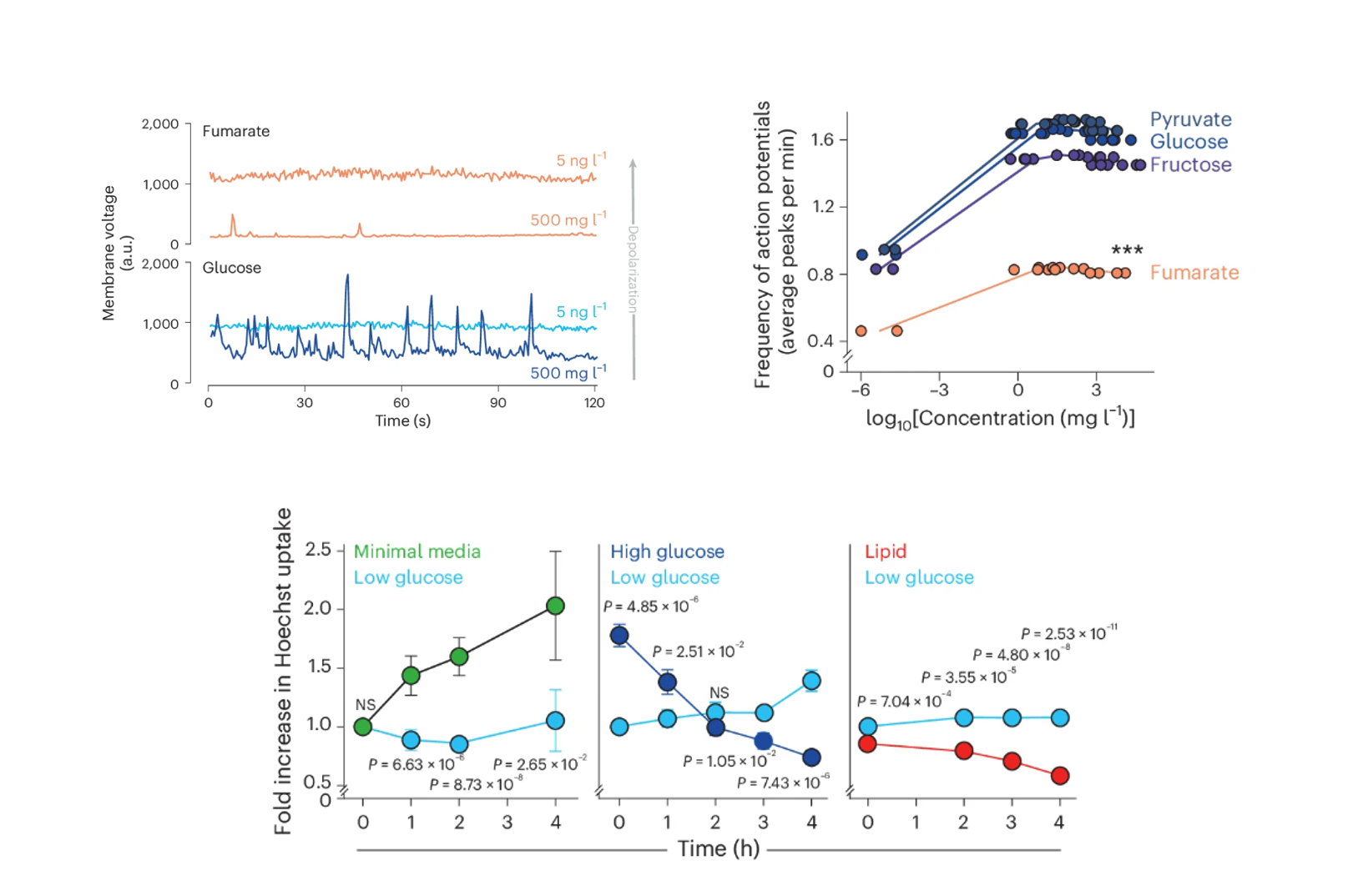
Figure 4. Metabolic state controls the frequency of membrane voltage oscillations and porin permeability across carbon sources.
Antibiotic consequences
Now the final question, why would you care about this? Well, it turns out that like sugars, many antibiotics rely on porin to get into the bacterium. Thus, the final phase of the project addressed whether the relationship between membrane voltage and uptake also applied to antibiotics, given that a significant portion of antibiotic resistance is due to changes in drug permeability. We focused on quinolones, a class of naturally fluorescent antibiotics, which made tracking their uptake in single cells easy. First, we confirmed that porins were the main route for quinolone entry, showing that porin knockouts had reduced uptake. The Kch knockout also showed decreased permeability, reinforcing the role of membrane potential. We then measured the EC50 of quinolones in both porin and Kch knockouts compared to wild-type cells, finding that both knockouts were more resistant to quinolones but not to colistin, an antibiotic that enters cells independently of porins. Finally, we revisited our ionophore experiments, treating cells with CCCP to see if altering the internal pH would affect ciprofloxacin uptake. Sure enough, we found that disrupting the proton gradient reduced antibiotic uptake. We hope this work helps explain why bacteria in certain metabolic states become naturally more resistant to drugs. It leaves us with a mechanistic bridge from metabolism to permeability. In lipid-rich environments—like certain intracellular niches—periplasmic acidification or slow bacterial “action potentials” likely close porins and reduce antibiotic entry. Therapeutically, activating Kch or otherwise depleting the proton gradient in the periplasm could, with adjutants, be a potential strategy to enhance antibiotic uptake and combat resistance (Figure 5).
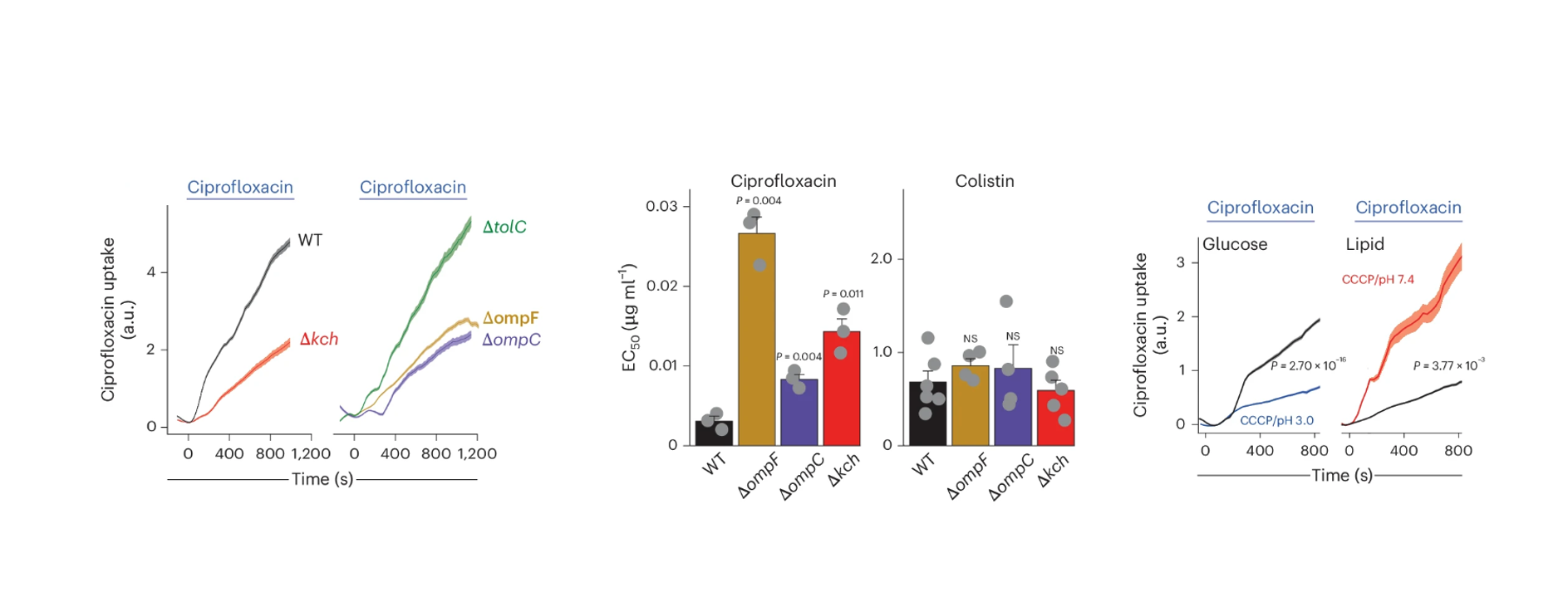
Figure 5. Porin and Kch control ciprofloxacin uptake and resistance, with proton gradient disruption dampening drug entry.
Few thank yous:
Before closing, just to note that this was a true team effort. Immense gratitude to Andres Floto—every success traces back to your ideas and leadership with Floto Lab. Grateful for the support from Cambridge University, the School of Clinical Medicine, and the Laboratory of Molecular Biology. Deep thanks to all co-authors: Stephen Trigg, Georgeos Hardo, Anja Hagting, Ieuan E. Evans, Chris Ruis, Ali F. Alsulami, David Summers, Felicity Crawshay-Williams, Tom L. Blundell, Lucas Boeck, and Somenath Bakshi.